- Szczegóły
-
Kategoria: Choroby rzadkie
-
Opublikowano: piątek, 22.09.2023, 17:21
-
Odsłony: 584
Informacja prasowa
ASMD to rzadka i postępująca choroba genetyczna, należąca do grupy lizosomalnych chorób spichrzeniowych, która występuje zarówno u dzieci, jak
i u dorosłych. Choroba ta do niedawna była całkowicie niewidzialna z uwagi na używanie zamierzchłej nomenklatury określającej ją jako chorobę Niemanna i Picka typu I oraz szereg podobnych objawów do choroby Gauchera. Dodatkowo pacjenci zmagający się z tą jednostką chorobową nie mieli żadnych możliwości terapeutycznych.

ASMD to rzadka i postępująca choroba genetyczna, należąca do grupy lizosomalnych chorób spichrzeniowych, która występuje zarówno u dzieci, jak
i u dorosłych. Choroba ta do niedawna była całkowicie niewidzialna z uwagi na używanie zamierzchłej nomenklatury określającej ją jako chorobę Niemanna i Picka typu I oraz szereg podobnych objawów do choroby Gauchera. Dodatkowo pacjenci zmagający się z tą jednostką chorobową nie mieli żadnych możliwości terapeutycznych.
– Zależy nam, aby w środowisku medycznym i w społeczeństwie ASMD przestało być niewidzialne. Wiemy, jak dużą wartość wnoszą działania edukacyjne wśród innych chorób rzadkich, dlatego wierzymy w pozytywne zmiany w świadomości również w tym obszarze. Jak wynika z sondy ulicznej, która została przeprowadzona w ramach kampanii „ASMD – ten typ tak ma. Był niewidzialny. A już leczyć się da” wiedza o chorobach rzadkich nadal jest na bardzo niskim poziomie. Czas to zmienić! Medycyna daje nam coraz więcej możliwości diagnostyki i leczenia, trzeba to wykorzystać. A bez wiedzy o istnieniu danej choroby odyseja diagnostyczna pacjenta może trwać latami – wyjaśnia Marzena Nelken, Dyrektorka Krajowego Forum Orphan, jedna z inicjatorek kampanii.
ASMD, czyli niedobór kwaśnej sfingomielinazy (z ang. acid sphingomyelinase deficiency) to rzadka i postępująca choroba genetyczna, która wiąże się ze znacznym odsetkiem śmiertelności, szczególnie wśród niemowląt i dzieci. Wielu pacjentów pediatrycznych nie dożywa wieku dorosłego. W organizmie chorego występuje brak lub niedobór enzymu kwaśniej sfingomielinazy, co w konsekwencji powoduje spichrzanie tłuszczu sfingomieliny w lizosomach komórek, prowadząc do wieloukładowych i wielonarządowych powikłań.
Szacuje się, że ASMD występuje z częstością 1 na 200 000 urodzeń.[1]
– Wyróżnia się trzy typy (postaci kliniczne) ASMD. Postać niemowlęca - typ A - to fenotyp
o wczesnym początku, w którym obserwuje się szybko postępującą neurodegenerację oraz zajęcie narządów wewnętrznych (znaczną hepatosplenomegalię, nawracające infekcje dróg oddechowych, małopłytkowość). Choroba prowadzi do śmierci zwykle przed ukończeniem przez dziecko 2-3 r.ż. ASMD typu B to postać przewlekła trzewna, w której początek objawów jest obserwowany w zróżnicowanym wieku, tj. od niemowlęctwa do dorosłości. Charakteryzuje się wolniejszą progresją zajęcia narządów wewnętrznych i brakiem objawów ze strony układu nerwowego. Postać przewlekła nerwowo-trzewna (ASMD typu A/B) stanowi fenotyp pośredni, który wyróżnia się późniejszym wystąpieniem objawów i wolniejszą progresją zajęcia ośrodkowego układu nerwowego niż w typie niemowlęcym – tłumaczy dr hab. n. med. Patryk Lipiński.
Jakie objawy powinny zaniepokoić?
Objawy związane z ASMD (deficytem kwaśnej sfingomielinazy) mogą być niewystarczająco wyraźne, aby wzbudzić podejrzenia lekarzy. Często diagnoza tej choroby opiera się na badaniach przesiewowych ze względu na różnorodność symptomów, jakie mogą występować. W konsekwencji prowadzi to do odysei diagnostycznej pacjentów.
– W przypadku ASMD charakterystyczne jest znaczne powiększenie objętości śledziony i/lub wątroby, które najczęściej nie powoduje żadnych dolegliwości, w związku z czym rozpoznanie choroby może nastąpić nawet po kilku latach. To stanowi pierwszy problem diagnostyczny, ponieważ lekarz pierwszego kontaktu czy pediatra, do którego zgłasza się pacjent, skieruje go w pierwszej kolejności do gastrologa lub hepatologa dziecięcego, w celu diagnostyki hepatosplenomegalii. Wykonanie podstawowych badań laboratoryjnych u pacjenta z ASMD - morfologii krwi obwodowej – pozwoli na stwierdzenie małopłytkowości (najczęściej), co sprawi, że pacjent zostanie odesłany do hematologa. Stwierdzenie podwyższonej aktywności aminotransferaz (tzw. prób wątrobowych) w korelacji z powiększeniem wątroby i/lub śledziony może sugerować pierwotną chorobę wątroby, co również opóźni diagnostykę
w kierunku ASMD – wyjaśnia dr Patryk Lipiński. – Do diagnostyki ASMD i tak naprawdę wszystkich wrodzonych chorób metabolicznych, trzeba podejść holistycznie – analizując obraz kliniczny w kontekście wyników badań dodatkowych. Niezmiernie ważna jest znajomość patofizjologii choroby – w przypadku ASMD obserwuje się gromadzenie sfingomieliny w makrofagach układu siateczkowo-śródbłonkowego, uszkodzenie hepatocytów oraz nacieczenie szpiku, odpowiadające za opisywane wyżej objawy – dodaje.
Najczęstszym, pierwszym objawem ASMD jest powiększenie śledziony i/lub wątroby, co wpływa negatywnie na ich funkcjonowanie. Powiększenie objętości śledziony występuje aż u ok. 90% badanych, a powiększenie wątroby u ok. 70% osób.[2]
U pacjentów pojawiają się także takie symptomy jak zmęczenie, powiększony obwód brzucha, oraz skłonność do siniaczenia z powodu zmniejszonej liczby płytek krwi.
Gromadzenie się sfingomieliny w pęcherzykach płucnych i ścianie oskrzeli może prowadzić do ograniczenia pojemności płuc i powoduje problemy z oddychaniem. U większości pacjentów z ASMD stwierdza się cechy choroby śródmiąższowej płuc.
ASMD – w cieniu innych chorób
W dawnej klasyfikacji chorób ASMD było określane jako choroba Niemanna-Picka typu I. Rozwój medycyny pozwolił na dokładną charakterystykę patofizjologii i rozróżnienie trzech postaci klinicznych ASMD. Aktualnie odchodzi się od nazewnictwa choroby Niemanna-Picka typu A, B oraz A/B i stosuje się terminologię ASMD (niedobór kwaśnej sfingomielinazy).
Pacjenci z ASMD mogą znajdować się w grupie pacjentów z podejrzeniem choroby Gauchera ze względu na podobne objawy, takie jak powiększenie śledziony i/lub wątroby oraz małopłytkowość. Dlatego ważne jest, aby przeprowadzić różnicową diagnozę, uwzględniając chorobę Gauchera. Aby odróżnić obie choroby, należy wykonać równoczesnie testy w kierunku ASMD oraz kwaśnej beta-glukozydazy (enzymu brakującego w chorobie Gauchera).
– U pacjentów z wyraźnie powiększoną śledzioną lub śledzioną i wątrobą, szczególnie
z towarzyszącą małopłytkowością, pierwszym krokiem powinno być wykluczenie/potwierdzenie choroby Gauchera, ze względu chociażby na jej częstsze występowanie (najczęstsza lizosomalna choroba spichrzeniowa), a następnie analiza aktywności kwaśnej sfingomielinazy w celu diagnostyki w wkierunku ASMD. Ponadto, w grupie pacjentów z objawami neurologicznymi (m.in. opóźnienie/regres psychoruchowy, objawy pozapiramidowe, ataksja), którym towarzyszy powiększenie wątroby i śledziony, należy wykluczyć/potwierdzić ASMD – tłumaczy dr Patryk Lipiński. – Rekomendowaną metodą diagnostyki ASMD jest analiza aktywności enzymu w leukocytach krwi obwodowej lub dostępnej od niedawna metodzie suchej kropli krwi (DBS) – dodaje.
Brak wiedzy wśród społeczeństwa
W ramach kampanii „ASMD – ten typ tak ma. Był niewidzialny. A już leczyć się da” została przeprowadzona sonda uliczna mająca na celu weryfikację wiedzy społeczeństwa z zakresu chorób rzadkich, a w tym niedoboru kwaśnej sfingomielinazy (ASMD).
– Pytając przechodniów o choroby rzadkie, duża liczba osób zdawała sobie sprawę, że jest ich więcej, niż sama nazwa wskazuje, ale bardzo często trudno im było wymienić konkretne jednostki chorobowe. Wiele z osób zwróciło uwagę, że za mało poświęca się tym ludziom czasu, że człowiek dopiero zdaje sobie sprawę z istnienia takiej choroby w momencie, kiedy ktoś bliski otrzyma taką diagnozę. Bardzo często podkreślano, że choroby rzadkie nie są tak nagłaśniane w telewizji – częściej słyszy się o tych powszechnych chorobach, szczególnie nowotworowych – zwraca uwagę Marzena Nelken.
– Uczestnicy sondy zostali zapytani także o to do czego, może prowadzić powiększenie śledziony i/lub wątroby – w tym pytaniu większość osób słusznie zwróciła uwagę na zaburzenie pracy całego organizmu, bóle brzuszne, a nawet śmierć. Powiększenie tych narządów jest bardzo charakterystycznym objawem w ASMD, na który chorują zarówno dzieci, jak i dorośli. Nie można lekceważyć tych symptomów, mimo że na początku mogą wydawać się błahe. Sam skrót choroby – ASMD – kojarzył się im z chorobą SMA, a nawet ze stwardnieniem rozsianym (SM). Dodatkowym problemem może być wcześniejsze nazewnictwo choroby określanej jako Niemann-Pick – zapewne jeszcze trochę czasu minie, zanim nowa nomenklatura ugruntuje się w świadomości lekarzy i społeczeństwa. To pokazuje nam jak ważne i jednocześnie trudne jest to, co robimy, jako stowarzyszenie, realizując różnego rodzaju kampanie. Choroby rzadkie w pewnym sensie żyją w cieniu chorób powszechnych, ale to nie znaczy, że Ci pacjenci zasługują na mniejszą uwagę – podkreśla Dyrektorka Krajowego Forum Orphan.
Link do sondy ulicznej: http://rzadkiechoroby.org/kampania-asmd/
Nowe możliwości dla pacjentów z ASMD
Przed 2022 rokiem pacjenci z ASMD byli leczeni jedynie objawowo. Sytuacja uległa zmianie
w 2022 roku, gdy Amerykańska Agencja ds. Żywności i Leków (FDA) oraz Europejska Agencja Leków (EMA) zatwierdziły do stosowania w leczeniu ASMD typu A/B i B olipudazę alfa. Dodatkowo FDA nadało temu lekowi status terapii przełomowej.
– Olipudaza alfa jest rekombinowaną ludzką kwaśną sfingomielinazą, stosowaną w leczeniu ASMD jako enzymatyczna terapia zastępcza, która zmniejsza nagromadzenie sfingomieliny mw narządach tj. śledzionie, wątrobie, płucach oraz w szpiku kostnym. Jest to pierwsza (ale skuteczna) terapia zatwierdzona do leczenia tej choroby. Enzymatyczna terapia zastępcza polega na substytucji brakującego enzymu – wyjaśnia dr Patryk Lipiński.
– Olipudaza alfa jest wskazana w leczeniu objawów ASMD typu B oraz A/B w niezwiązanych
z zajęciem ośrodkowego układu nerwowego, ponieważ enzym nie przenika bariery krew-mózg, tym samym nie ma wpływu na objawy neurologiczne – dodaje.
– Jest wiele chorób rzadkich, w których pacjenci leczeni są jedynie objawowo z uwagi na brak terapii. Tym bardziej zawsze cieszy nas moment, w którym sytuacja ulega zmianie i rozwój medycyny pozwala na skuteczne leczenie. ASMD może ujawnić się w różnym wieku. Im szybciej zostanie zatrzymany rozwój choroby tym większa korzyść dla pacjenta, szczególnie dla tych młodych pacjentów, którzy mają przed sobą całe życie – puentuje Marzena Nelken, inicjator kampanii „ASMD – ten typ tak ma. Był niewidzialny. A już leczyć się da”.
Piśmiennictwo
[1] Lipiński P., Tylki-Szymańska A., Deficyt kwaśnej sfingomielinazy – obraz kliniczny, diagnostyka i leczenie, Standardy Medyczne/Pediatria 2023, T. 20, 159-164
[2] McGovern MM, Avetisyan R, Sanson BJ i wsp. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J Rare Dis 2017
Informacja prasowa
GdL 9/2023
- Szczegóły
-
Kategoria: Choroby rzadkie
-
Opublikowano: wtorek, 27.06.2023, 16:57
-
Odsłony: 620
Nocna napadowa hemoglobinuria (w skrócie PNH) jest przewlekłą, postępującą chorobą ultrarzadką. Jej występowanie szacuje się na 1,2 na milion osób rocznie. PNH dotyka głównie ludzi młodych – mediana wieku zachorowania to ok. 30 lat.
Choroba ma podłoże genetyczne; charakteryzuje się wewnątrznaczyniową hemolizą (rozpadem czerwonych krwinek w organizmie chorego), która jest spowodowana zaburzeniami mechanizmu odpowiedzi immunologicznej, tj. ciągłym stanem aktywacji układu dopełniacza.
Objawy kliniczne obejmują m.in. zakrzepicę i niewydolność nerek, które mogą być przyczyną przedwczesnego zgonu. Ok. 35% nieleczonych pacjentów umiera z powodu PNH w ciągu 5 lat od postawienia diagnozy. Ponadto, chorzy doświadczają wielu innych objawów, tj. nadciśnienie płucne, niedokrwistość, duszność, przewlekłe zmęczenie, hemoglobinuria, zaburzenia połykania, wzmożone napięcie mięśni, bóle brzucha, zaburzenia erekcji. W znaczący sposób wpływają one na jakość życia, uniemożliwiając chorym normalne funkcjonowanie.
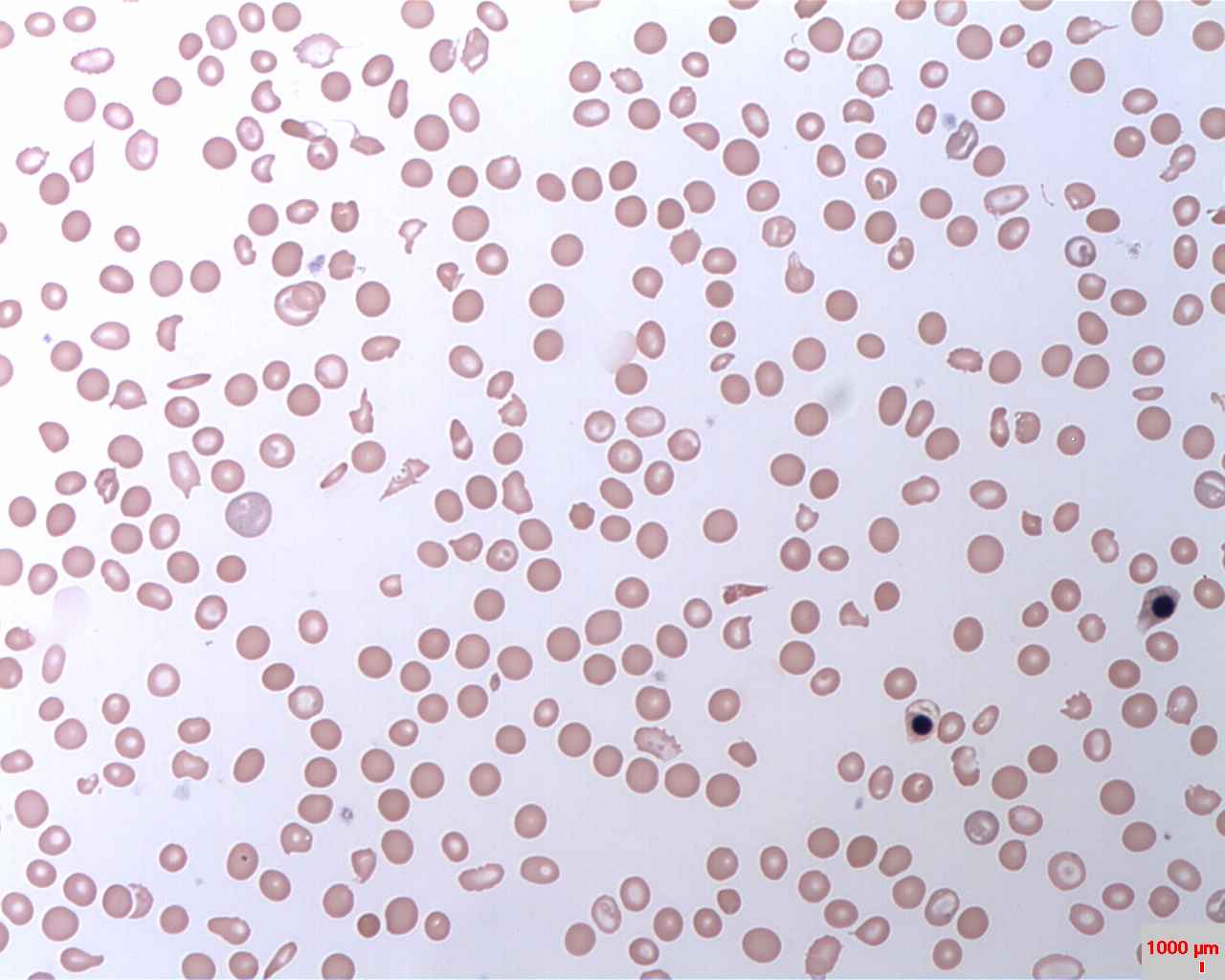
Wewnątrznaczyniowa niedokrwistość hemolityczna
Źródło: https://en.wikipedia.org/wiki/Paroxysmal_nocturnal_hemoglobinuria
W Polsce aktualnie zdiagnozowanych jest ok. 100 pacjentów z PNH. Pod koniec czerwca 2023 roku opracowano raport „Sytuacja chorych na nocną napadową hemoglobinurię w Polsce”.
– Pacjenci z PNH to ludzie młodzi, którzy chcą normalnie pracować, podróżować, marzyć, korzystać i cieszyć się z życia. Niestety, życie z tą chorobą jest trudne, a czasem nawet nie do zniesienia. Raport potwierdza, że PNH, ale również jej leczenie, wpływa na każdy aspekt codzienności pacjenta i jego bliskich: począwszy od edukacji i pracy, przez sytuację finansową, po plany na przyszłość związane na przykład z założeniem rodziny.
Jak wynika z raportu, wpływ choroby na sferę emocjonalną pacjentów może być równie niszczycielski, jak jej skutki fizyczne. Jeszcze przed zdiagnozowaniem PNH, chorzy czują się niezrozumiani przez otoczenie, a nawet stygmatyzowani, ponieważ wskutek najczęstszych objawów choroby, jakimi są brak energii czy szybka męczliwość, są postrzegani jako osoby leniwe i mało ambitne.
Proces ustalenia rozpoznania w przypadku ponad 1/3 badanych trwał powyżej 1. roku, a 1 na 10 pacjentów otrzymało prawidłową diagnozę dopiero po 3 latach od pojawienia się pierwszych objawów.
Przełom w leczeniu chorych z PNH nastąpił w 2007 roku, kiedy w Europie zarejestrowano pierwszy lek na tę chorobę, ekulizumab (inhibitor C5 układu dopełniacza), który od 2018 roku jest dostępny dla pacjentów w Polsce. W ciągu kilku ostatnich lat wachlarz terapii PNH rozszerzył się o dwa innowacyjne leki, tj. rawulizumab (pierwszy długodziałający inhibitor C5 układu dopełniacza) i pegcetacoplan (inhibitor C3 układu dopełniacza, stosowany u pacjentów z PNH nieodpowiadających optymalnie na leczenie inhibitorem C5), których udostępnienie wpłynęłoby na optymalizacje leczenia pacjentów z tą chorobą.
Nowe terapie są mniej obciążające dla chorych niż aktualnie stosowane leczenie. Przykładowo, podawany dożylnie rawulizumab wymaga stosowania co 8 tygodni, a więc aż czterokrotnie rzadziej niż ekulizumab, co oznacza 6-7 podań rocznie zamiast 26 podań, a jest równie skuteczny, co obecnie dostępny lek.
Z zawartej w raporcie analizy wynika, że zmniejszenie częstości podań leku zredukuje obciążenie chorobą pacjentów, przełoży się na poprawę jakości ich życia rodzinnego, umożliwi większą aktywności zawodową i społeczną chorych oraz ich bliskich, a także przyniesie zyski dla systemu ochrony zdrowia. – Udostępnienie terapii podawanych rzadziej pozwoli na osiągnięcie korzyści systemowych poprzez redukcję obciążenia personelu medycznego oraz kosztów związanych z wizytami i podaniem leku.
Ponadto, biorąc pod uwagę skalę problemów psychicznych zgłaszanych przez pacjentów biorących udział w badaniu, dla pełnej poprawy sytuacji chorych z PNH w Polsce niezbędne jest również systemowe wsparcie psychologiczne, np. w postaci opieki psychologicznej dostępnej w każdym ośrodku hematologicznym zajmującym się leczeniem PNH.
Źródło: Informacja prasowa (Oprac. K.K.)
GdL 6/2023
- Szczegóły
-
Kategoria: Choroby rzadkie
-
Opublikowano: piątek, 03.02.2023, 10:00
-
Odsłony: 1143
Wywiad z dr n. med. Agnieszką Kułagą, specjalistą neurologiem z Oddziału Neurologii Krakowskiego Szpitala Specjalistycznego im. Jana Pawła II
Co wiadomo już dzisiaj na temat miastenii? Jak można scharakteryzować tę chorobę?
Miastenia jest nabytą chorobą autoimmunologiczną, w przebiegu której dochodzi do produkcji przeciwciał skierowanych przeciwko białkom złącza nerwowo-mięśniowego. W 80 proc. przypadków są to przeciwciała skierowane przeciwko receptorom dla acetylocholiny (AChR-Ab, ARAB), 10 proc. pacjentów ma przeciwciała skierowane przeciwko mięśniowo swoistej kinazie tyrozyny, czyli tzw. przeciwciała anty-MuSK. Kluczowym objawem choroby jest męczliwość mięśni szkieletowych, czyli narastanie osłabienia mięśni w trakcie ich aktywacji. Drugą typową cechą miastenii, wynikającą z męczliwości mięśni, jest zmienność objawów, zarówno w ciągu dnia, jak i dni, tygodni, czy nawet miesięcy.
Nie wiadomo, co prowadzi do rozwoju tej choroby. Najczęściej czynnikiem wyzwalającym wystąpienie objawów jest infekcja. Natomiast w patogenezie miastenii bierze się pod uwagę zaburzenia układu immunologicznego, dysregulację hormonalną, wpływ czynników środowiskowych oraz predyspozycje genetyczne.
Kto choruje na miastenię? Czy wiek, płeć, status socjoekonomiczny odgrywają w przypadku tej choroby jakąś rolę? Czy można mówić o grupie zwiększonego ryzyka zachorowania na miastenię?
Na miastenię można zachorować w każdym wieku. Chorują na nią zarówno dzieci, jak i osoby dorosłe. Możemy wyróżnić grupy pacjentów w zależności od wieku, w którym wystąpiły u nich objawy choroby. Na miastenię o wczesnym początku chorują osoby przed 50. rokiem życia, trzy razy częściej są to kobiety. Natomiast na miastenię o późnym początku chorują osoby po 50. roku życia, w tym wypadku nie ma różnicy w częstości zachorowania między kobietami i mężczyznami. Jest też grupa pacjentów o bardzo późnym początku zachorowania – należą do niej osoby powyżej 65. roku życia. Średni wiek zachorowania na miastenię w Polsce wynosi 61 lat. Zapadalność, czyli liczba nowych pacjentów przed 50. rokiem życia, którzy zachorują w danym roku, wynosi 1 na 100 tysięcy mieszkańców, ale już po 60. roku życia liczba ta wzrasta do 5. Jeżeli mówimy o chorobowości, to w Polsce na miastenię chorują średnio 22 osoby na 100 tysięcy mieszkańców. Jednak kiedy spojrzymy na wiek chorych, to okazuje się, że między 70.-90. rokiem życia choruje około 70 osób na 100 tysięcy populacji.
Status socjoekonomiczny nie ma wpływu na zachorowanie na miastenię, nie istnieje również grupa osób o zwiększonym ryzyku rozwoju miastenii. Możemy natomiast zakładać, że osoba chorująca na jedną chorobę autoimmunologiczną ma zwiększone ryzyko rozwoju drugiej takiej choroby, w tym miastenii.
Ile osób choruje na miastenię w Polsce?
Według najnowszych danych epidemiologicznych, opracowanych przez prof. Annę Kosterę-Pruszczyk wraz z zespołem, w Polsce choruje około 9 tysięcy osób.
Czy miastenia daje jakieś charakterystyczne objawy? Na co powinni zwrócić uwagę lekarze, zwłaszcza podstawowej opieki zdrowotnej, do których pacjenci trafiają najczęściej? Do jakiego specjalisty powinien zostać skierowany pacjent, u którego lekarz POZ podejrzewa miastenię?
Najczęstszymi objawami występującymi u pacjentów z miastenią są objawy oczne – opadanie powiek, zazwyczaj asymetryczne, oraz podwójne widzenie. U chorego na miastenię mogą również wystąpić inne objawy, takie jak: zaburzenia mowy, gryzienia i połykania czy czterokończynowe osłabienie mięśni. Im dłużej pacjent ogląda telewizję lub na przykład coś czyta, tym widziany przez niego obraz robi się coraz bardziej niewyraźny i staje się podwójny. Im dłużej mówi, tym jego mowa staje się mniej zrozumiała i sepleniąca. Pacjent zaczyna jeść i z czasem obserwuje, że nie jest już w stanie przełknąć kolejnej porcji. Zaczyna wchodzić po schodach i po jakimś czasie nie może pokonać następnego stopnia. Po odpoczynku te objawy najczęściej zmniejszają swoje nasilenie, mogą być minimalne. Jak już powiedziałam, nasilenie objawów jest zmienne. Często bywa tak, że rano pacjent budzi się wypoczęty, ma niewielkie objawy, ale już w godzinach popołudniowych stają się one wyraźne i utrudniają mu funkcjonowanie. Jednak zdarza się również, że objawy są najbardziej odczuwalne w godzinach porannych i z czasem maleją. Każdy pacjent choruje inaczej.
Objawem, który powinien wzbudzić u lekarza podstawowej opieki zdrowotnej podejrzenie miastenii, jest asymetryczne opadanie powiek oraz podwójne widzenie. Może być i tak, że pacjent zgłasza się do lekarza bez żadnych objawów, ale o nich mówi. W takim przypadku należy zwrócić uwagę na tę zmienność objawów, o której pacjent opowiada, dopytać go, czy podobne objawy występowały u niego w przeszłości, kiedy są najbardziej nasilone i czy ich początek nie nastąpił po infekcji. Lekarz rodzinny, który ma przed sobą pacjenta bez objawów albo z bardzo mało nasilonymi objawami, może podjąć próbę zmęczenia go. W jaki sposób? Na przykład poprosić pacjenta, żeby głośno policzył do 50, dać mu jakiś dłuższy tekst do przeczytania, poprosić go, żeby robił przysiady, otwierał i zamykał oczy. Jeżeli pacjent wykonuje takie ćwiczenia, to istnieje bardzo duże prawdopodobieństwo, że wystąpią u niego objawy miastenii. W przypadku wysunięcia podejrzenia tej choroby, pacjent powinien zostać skierowany jak najszybciej do specjalisty neurologa.
Porozmawiajmy teraz o diagnostyce miastenii. Co jest podstawą rozpoznania tej choroby? Jakie badania trzeba wykonać, aby upewnić się, że pacjent rzeczywiście ma miastenię? Jak długo czeka się na diagnozę, od czego zależy ten okres oczekiwania?
Tak jak już powiedziałam, aby wysunąć podejrzenie miastenii u pacjenta, powinien on mieć typowe objawy, czyli męczliwość mięśni oraz zmienność objawów. Jeżeli pacjent spełnia kryteria kliniczne, to w pierwszej kolejności powinien mieć oznaczony poziom przeciwciał. Najpierw oznacza się poziom przeciwciał przeciwko receptorom dla acetylocholiny, a w następnej kolejności przeciwciała anty-MuSK. Jeżeli pacjent ma typowe objawy i stwierdza się u niego obecność przeciwciał, to możemy już u niego rozpoznać miastenię. Wyzwanie stanowią chorzy seronegatywni, którzy wymagają poszerzenia diagnostyki.
U każdego chorego należy wykonać badanie tomografii komputerowej klatki piersiowej w poszukiwaniu patologii grasicy. Wykonuje się również testy elektrofizjologiczne, czyli elektrofizjologiczną próbę nużliwości, a w wątpliwych przypadkach także badanie EMG pojedynczego włókna mięśniowego (SFEMG).
Okres oczekiwania na diagnozę zależy od tego, jak szybko pacjent trafi do lekarza neurologa. Najczęściej rozpoznanie choroby występuje w drugim roku od wystąpienia pierwszych objawów. Ale zdarzają się również chorzy, u których postawienie rozpoznania zajęło kilka lat, ponieważ zostali skierowani do nieodpowiedniego specjalisty. Pacjenci mówiący o objawach, a nie mający ich, są czasem kierowani w pierwszej kolejności do psychiatry z podejrzeniem zaburzeń psychosomatycznych. Zdarzają się też chorzy, którzy zostali zoperowani z powodu zeza. Na szczęście takich przypadków jest coraz mniej, bo zwiększa się świadomość tej choroby i pacjenci najczęściej są kierowani bezpośrednio do neurologa.
Jaki wpływ na dalsze postępowanie diagnostyczno-terapeutyczne ma stwierdzenie u pacjenta obecności przeciwciał przeciwko receptorom dla acetylocholiny lub obecności przeciwciał przeciwko białku MuSK?
Obraz kliniczny pacjentów, którzy mają przeciwciała przeciwko receptorom dla acetylocholiny i pacjentów z przeciwciałami anty-MuSK różni się. Osoby z przeciwciałami anty-MuSK zazwyczaj chorują ciężej, mają objawy zlokalizowane głównie w obrębie twarzy i opuszki, i często występuje u nich zanik języka. Tacy chorzy nie reagują na standardowe leczenie objawowe bromkiem pirydostygminy i mają nasilone działania niepożądane po tym leku. W tej grupie zazwyczaj bardzo wcześnie wprowadza się od leczenie immunosupresyjne.
Jaki przebieg ma miastenia i co to jest przełom miasteniczny?
Każdy pacjent choruje inaczej. Są osoby, które mają łagodne objawy i wymagają jedynie leczenia objawowego. Ale są też pacjenci, którzy mają bardzo nasilone objawy i u których stosuje się kilka leków.
Przełom miasteniczny jest stanem zagrożenia życia, który występuje u około 15 proc. pacjentów z miastenią. Najczęściej rozwija się w ciągu dwóch pierwszych lat od wystąpienia pierwszych objawów. Z reguły jest wyzwalany przez infekcję. Częściej występuje u pacjentów z nasilonymi objawami opuszkowymi. Jest definiowany jako wystąpienie objawów niewydolności oddechowej, wynikającej z osłabienia przepony i dodatkowych mięśni oddechowych. Pacjenci w przełomie miastenicznym wymagają hospitalizacji woddziale intensywnej terapii medycznej, podłączenia do respiratora lub zastosowania nieinwazyjnej wentylacji mechanicznej. Przełom miasteniczny leczy się u pacjentów wlewami dożylnymi immunoglobulin lub zabiegami plazmaferezy.
Jakie są standardy leczenia miastenii? Czy w przypadku tej choroby można mówić o terapii spersonalizowanej?
Są opracowane wytyczne mówiące o sposobie leczenia pacjentów z miastenią. Dla każdego chorego leczenie dopiera się indywidualnie, biorąc pod uwagę jego wiek, choroby współistniejące, a także styl życia, jaki prowadzi. Każdy pacjent ma inne oczekiwania dotyczące efektów leczenia i to również należy brać pod uwagę. Wiadomo, że inaczej będziemy leczyć pacjentów młodych, aktywnych, bez chorób współistniejących, a inaczej osoby w starszym wieku z licznymi obciążeniami, a jeszcze inaczej kobiety w ciąży.
Czy dostępne leczenie to leczenie wpływające na przebieg choroby czy jedynie łagodzące jej objawy?
Nie istnieje lek, który może wyleczyć z miastenii. Do wyboru mamy leki łagodzące objawy. W Polsce dostępny jest aktualnie jeden lek z tej grupy – bromek pirydostygminy, inhibitor acetylocholinoesterazy. Drugą grupę stanowią leki modulujące przebieg choroby – sterydy oraz leki immunosupresyjne. Ich celem jest zmniejszenie odpowiedzi immunologicznej i wprowadzenie choroby w remisję.
Jak duża jest grupa pacjentów, u których objawy pomimo leczenia nie są kontrolowane lub są kontrolowane niewystarczająco? Jakie są opcje postępowania w przypadku takich pacjentów?
U około 75 proc. pacjentów po wprowadzeniu leczenia lekami objawowymi i immunomodulującymi objawy choroby są dobrze kontrolowane, to znaczy objawy albo w ogóle nie występują, albo mają minimalne nasilenie i nie wpływają istotnie na jakość życia. Z kolei u około 15 proc. chorych, pomimo leczenia objawy są nasilone, nie udaje się wprowadzić chorego w remisję lub objawy u niego nawracają przy próbach redukcji dawek leków immunosupresyjnych. W tej grupie są również pacjenci, którzy mają nasilone działania niepożądane sterydów bądź leków immunosupresyjnych oraz ci, którzy nie tolerują tych leków. Tacy chorzy stanowią wyzwanie dla neurologów. W ich przypadku dobiera się kombinacje różnych leków i próbuje się stosować terapie ratunkowe, na przykład rytuksymabem lub ekulizumabem. Czasami tacy chorzy wymagają cyklicznego podawania immunoglobulin dożylnie, czy wykonywania regularnych zabiegów plazmaferezy.
Na szczęście w ostatnich latach wiele się dzieje w miastenii. Pojawiają się nowe leki, których mechanizm jest ukierunkowany na konkretne molekuły układu immunologicznego. Mówimy o lekach, które dezaktywują układ dopełniacza, wpływają na limfocyty B lub blokują noworodkowy receptor Fc (FcRn). W przypadku kilku leków wykazano już bezpieczeństwo i co najważniejsze, skuteczność działania w miastenii. Aktualnie są one dostępne dla pacjentów w ramach badań klinicznych, które są prowadzone w wielu ośrodkach w Polsce. Mam nadzieje, że za kilka lat będą one w Polsce szerzej dostępne i zarejestrowane do leczenia pacjentów z miastenią.
Jak często zalecane są wizyty kontrolne?
Częstotliwość wizyt kontrolnych zależy od stanu pacjenta. Są chorzy wymagający częstych kontroli, nawet raz na dwa tygodnie albo raz na miesiąc, ale i tacy, którzy przychodzą na kontrolę raz na sześć miesięcy czy nawet tylko raz na rok. Ważne jest, aby chory miał świadomość, że w przypadku pogorszenia może w każdej chwili zgłosić się czy zadzwonić do swojego neurologa prowadzącego.
Czy w Polsce są ośrodki wyspecjalizowane w leczeniu miastenii? Ile ich jest i w jakich miastach?
W każdym województwie znajduje się jeden, a czasem kilka ośrodków specjalizujących się w diagnozowaniu i leczeniu pacjentów z miastenią, więc pacjenci nie powinni mieć problemów ze znalezieniem takiego ośrodka. Najbardziej opiniotwórcza, jeśli chodzi o choroby nerwowomięśniowe, jest Klinika Neurologii Warszawskiego Uniwersytetu Medycznego pod kierownictwem prof. Anny Kostery-Pruszczyk.
Dla wielu chorych miastenia nie jest jedyną chorobą, z którą muszą się zmagać. Czy leczenie chorób współistniejących może nasilić u nich objawy miastenii?
Pacjent z miastenią może mieć także inne choroby, wymagające leczenia. Wszystkie choroby współistniejące powinny być leczone zgodnie z aktualnymi wytycznymi. Nie należy bać się miastenii. Szczególnie ważne jest wczesne leczenie infekcji i dobra kontrola hormonów tarczycowych u chorych ze współistniejącą chorobą tarczycy. Należy również zwrócić uwagę na choroby, które mogą rozwinąć się w przebiegu przewlekłego stosowania sterydów i wcześnie rozpocząć ich leczenie. Pacjenci leczeni sterydami powinni być kontrolowani pod kątem rozwoju zaburzeń lipidowych, cukrzycy, zaćmy, nadciśnienia tętniczego, osteoporozy, choroby wrzodowej żołądka. Dobra kontrola i odpowiednie leczenie chorób współistniejących ma istotny wpływ na przebieg miastenii.
Jakich leków nie powinni przyjmować chorzy na miastenię?
Nie ma leków, które są bezwzględnie przeciwwskazane u chorych na miastenię. Jeszcze kilka lat temu istniała dosyć długa lista leków, których nie należało stosować w miastenii, ale aktualnie wytyczne są coraz bardziej liberalne. Są leki, które mogą, ale nie muszą nasilać objawów miastenii. Jeśli pacjent ma silne wskazania do ich zastosowania, to powinien je otrzymać. Trzeba tylko pamiętać, że mogą one nasilać objawy miastenii i w takich przypadkach należy skonsultować się z lekarzem prowadzącym.
Do leków, które mogą nasilać objawy miastenii zaliczamy niektóre antybiotyki (np. makrolidy, aminoglikozydy i fluorony), leki stosowane w leczeniu chorób kardiologicznych (np. werapamil), leki stosowane w znieczuleniu ogólnym (np. pankuronium), czy leki stosowane w psychiatrii (np. lit). Grupy leków, które mogą nasilić objawy miastenii są powszechnie dostępne w internecie.
Jaka jest rola diety w leczeniu miastenii? Czy są jakieś produkty, których chorzy na miastenię powinni unikać?
Nie ma diety specjalnie polecanej dla chorych na miastenię, nie istnieje coś takiego jak dieta miasteniczna. Generalnie pacjenci powinni odżywiać się zdrowo, czyli stosować dietę śródziemnomorską, która jest zalecana właściwie dla każdego. Chorzy, którzy mają inne choroby współistniejące, np. chorzy z zaburzeniami lipidowymi, powinni stosować dietę niskotłuszczową, chorzy na cukrzycę – dietę cukrzycową itd. Nie ma produktów czy pokarmów, które są przeciwwskazane w miastenii.
W jaki sposób rodzina i bliscy mogą wesprzeć chorego na miastenię w radzeniu sobie z chorobą i kontrolowaniu jej?
Bardzo ważne jest, aby pacjent miał poczucie, że rodzina oraz jego najbliżsi akceptują chorobę i ją rozumieją. Czasami pacjenci opowiadają, że po powrocie z pracy, w której spędzają cały dzień, mają już tak nasilone objawy miastenii, że muszą odpocząć, poleżeć, przespać się. A od członków swojej rodziny słyszą czasem, że udają, są leniwi, mogli pracować cały dzień, a w domu to muszą leżeć. Do takich sytuacji w ogóle nie powinno dochodzić, bo chorzy z miastenią niczego nie udają. Oni naprawdę są zmęczeni, nie mogą poruszać kończynami, mówić, przełykać. Najbliżsi powinni im okazywać wsparcie i zrozumienie, zachęcać ich do odpoczynku, a nie ganić ich za to, że chcą odpocząć.
Jakie są największe wyzwania i potrzeby pacjentów chorujących na miastenię z perspektywy lekarza?
Na pewno największym wyzwaniem jest wczesna diagnostyka i wczesne rozpoczęcie leczenia. Dużo pacjentów zgłaszających się do neurologa ma jakąś wiedzę na temat miastenii, zaczerpniętą z Internetu. Wiemy, że często te informacje są nieprawdziwe, dlatego najważniejszym zadaniem dla lekarza jest dokładne wytłumaczenie choremu, na czym polega ta choroba, jak się ją leczy, jakich efektów leczenia należy się spodziewać, na jak długo jest ono zaplanowane i jak ta choroba wpływa na życie codzienne. Istnieją stowarzyszenia pacjentów, np. bardzo aktywnie działające Polskie Stowarzyszenie Chorych na Miastenię Gravis „Gioconda”. Na stronie stowarzyszenia można dowiedzieć się na temat samej choroby, pacjenci wzajemnie się wspierają i dzielą się swoimi historiami choroby.
Ważne jest również, aby pacjent wiedział, w jakich sytuacjach musi bezwzględnie skontaktować się ze swoim lekarzem neurologiem i kiedy powinien zgłosić się do szpitala z powodu nasilenia objawów. Nasilone objawy opuszkowe (mowy, gryzienia, przełykania) są bezwzględnym wskazaniem do hospitalizacji w oddziale neurologii.
Pacjenci muszą sobie również zdawać sprawę z tego, jak ważne jest leczenie chorób współistniejących i wczesne rozpoczęcie leczenia infekcji. Muszą o siebie dbać. Kiedy widzą, że mają infekcję, powinni zostać w domu i zażywać leki przepisane przez lekarza podstawowej opieki zdrowotnej. Im wcześniej rozpoczną leczenie infekcji i chorób współistniejących, tym mniejsze jest ryzyko, że dojdzie do nasilenia objawów miastenii
rozmawiała
Maja Marklowska - Tomar
GdL 2/2023




